|
Introduction
LPS
is a constituent of the outer membrane of gram-negative
bacteria and evokes an inflammatory response by activation of monocytes and
endothelial cells. LPS-induced cellular responses are the net result
of the interaction of LPS with various plasma components such as soluble CD14,
LPS-binding protein (LBP) and membrane receptors such as membrane-bound CD14
and Toll-like receptors. This initiation of cellular responses
is essential for the
host defense against bacterial infections. However, if large amounts
of endotoxin are present in the circulation, an excessive cellular response can
be deleterious for the host, and, therefore, endotoxin-inactivating
processes are of extreme
importance.
LPS
is detoxified in the circulation by incorporation into
lipoproteins (reviewed in ref. 1). Physiological levels of lipoproteins
protect against endotoxicity in vitro and in vivo (2, 3). Early studies have demonstrated an
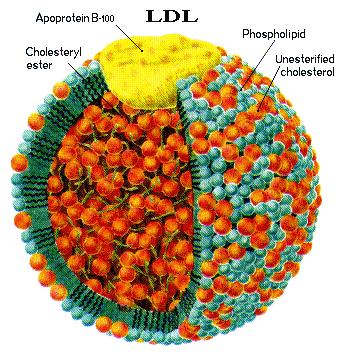
interaction of LPS with HDL (4); albeit later, also VLDL and LDL were found
to bind and inactivate LPS (5–7). Consistent with this, LDL, VLDL,
chylomicrons, and HDL all have been observed to reduce the lethal effect of
endotoxin in mice (8–10).
Evidence for
a physiological role for LBP in inflammation
is supported by studies that demonstrate enhanced mortality and uncontrolled
multiplication and spread of bacteria in LBP knockout mice compared with
wild-type mice after intraperitoneal administration of bacteria (11). The results of these studies indicate that LBP
is required to induce a rapid inflammatory response, which is essential for the
resistance to bacteria. However, LBP has the paradoxical dual function of
sensitizing the immune system to endotoxin and, on the other hand, enhancing
detoxification of endotoxin. LBP catalyzes the transfer of LPS into
lipoproteins, thereby enhancing LPS detoxification (12). Likewise, LBP catalyzes the lipoprotein
neutralization of lipoteichoic acid, a component of the cell membrane of
gram-positive bacteria (13). Lamping et al. demonstrated in a murine model
that high levels of LBP in the circulation, as seen during an acute-phase
response, inhibit LPS effects and prevent mortality induced by endotoxemia (14). The latter observation strongly supports a
physiological role for LBP-dependent detoxification of LPS in the host defense.
Endotoxemia
induces an acute-phase response characterized
by multiple physiological adaptations. This response appears to play a role in
host defense mechanisms, although its physiological relevance needs further
elucidation. One aspect of the acute-phase response is a dramatic rise in
circulating levels of LBP (15). Concomitantly, large changes in serum lipid and
lipoprotein concentrations occur. Circulating levels of total cholesterol, LDL
cholesterol, and HDL cholesterol decrease, whereas serum triglyceride and VLDL
levels increase (16). In addition, alterations in apolipoprotein
levels are observed (16, 17). ApoA-I concentrations drop, and HDL becomes
depleted in apoA-I (16, 18). In contrast, apoB levels are not affected by
either viral or bacterial infection (16).
Others found
evidence for an association of
LBP with apoA-I–containing lipoproteins in plasma from healthy persons (12). We consider that the physical association of LBP
with these lipoproteins may be important for the cooperation of LBP and
lipoproteins in the detoxification of endotoxin. However, the strong reduction
of apoA-I and HDL levels that coincides with the dramatic raise in LBP levels
during endotoxemia seems in contrast with this cooperative function. Because it
is firmly established that LDL and VLDL are critical in the survival of
infection with gram-negative bacteria (19) and that circulating levels of these lipoproteins
are relatively high during inflammation compared with HDL levels, the present
study was undertaken to investigate whether LBP associates with LDL and VLDL.
To this end, the distribution of LBP among lipoproteins was studied in serum of
healthy and septic persons. Subsequently, we investigated the effect of the
association of LBP with lipoproteins and apolipoproteins on the LPS-binding
capacity.
Go to:
Methods
Reagents.
LPS
from Escherichia
coli, serotype 055:B5, was purchased from Sigma Chemical Co. (St. Louis,
Missouri, USA). Purified apoA-I and apoB were derived from Calbiochem (La
Jolla, California, USA) and ICN Radiochemicals Inc. (Irvine, California, USA),
respectively. Polyclonal antibodies to human LBP were obtained by immunizing
rabbits with purified human LBP. Protein A–purified anti-LBP IgG was biotinylated
following standard procedures. Anti-human LBP mAb HM14 was obtained by
immunizing mice with LBP following classical procedures. Horseradish
peroxidase–labeled mAb against apoA-I and apoB were gifts from L. Sorell
(Center for Genetic Engineering and Biotechnology, Havana, Cuba).
Blood samples.
Blood was collected
from healthy donors and
from septic patients. Informed consent was obtained from healthy donors and
relatives of septic patients. To prepare serum, blood was allowed to clot for 2
hours at room temperature. Serum was separated by centrifugation and stored at
–80°C until use. Fresh serum was used for isolation of lipoprotein fractions.
Lipid electrophoresis and Western blot analysis.
Agarose electrophoresis
was performed in
barbital buffer (pH 8.6) using the Paragon Lipoprotein Electrophoresis kit P/N
(Beckman Instruments, Brea, California, USA) according to the manufacturer’s
instructions. Agarose gel electrophoresis was followed by electrophoretic
transfer onto an Immobilon-P membrane (Millipore Corp., Bedford, Massachusetts,
USA) in blotting buffer (25 mM TRIS, 192 mM Glycine, 10% methanol) using the
Phast system (Pharmacia Biotech A/B, Uppsala, Sweden). After transfer, the
membranes were blocked with 1% BSA in PBS for 1 hour at 37°C and washed with
0.1% BSA 0.5% Tween in PBS. For detection of LBP, membranes were incubated with
biotin-labeled polyclonal antibody specific for human LBP, washed, and
incubated with peroxidase-labeled streptavidin. After detection of LBP, apoA-I
was probed on the same blot after elution of the LBP antibodies by incubation
with 0.1 M Glycine-HCl buffer (pH 2.5) for 15 minutes at 37°C. Localization of
apoA-I was followed by detection of apoB in a similar protocol. For
immunodetection of apoA-I and apoB, peroxidase-labeled mAb’s were used.
Interactions of LPS with serum lipoproteins were studied by preincubation of
biotin-labeled LPS with human serum overnight at 37°C. After preincubation,
serum was subjected to electrophoresis and blotted and membranes were incubated
with peroxidase-labeled streptavidin. To study LPS binding to isolated
(apo)lipoproteins, human serum was first subjected to electrophoresis and
blotted and membranes were incubated with biotinylated LPS, followed by
incubation with peroxidase-labeled streptavidin (Zymed Laboratories Inc., South
San Francisco, California, USA). Peroxidase activity was detected by
chemiluminescent substrates (SuperSignal Substrate, Pierce Chemical Co.,
Rockford, Illinois, USA) according to the manufacturer’s recommendation.
Lipoprotein fractionation.
Lipoproteins
were isolated from pooled fresh
normal human serum (six healthy donors) by a 22-hour single spin density
gradient ultracentrifugation at 200,000g and 17°C according to
Terpstra et al. (20), using a Beckman XL-80 ultra centrifuge with a
SW40 rotor and ultraclear centrifuge tubes (Beckman Instruments). A step
gradient was constructed from 2 ml serum adjusted to density 1.250 g/ml with
KBr and sucrose; two NaCl/KBr solutions with density 1.225 and 1.100 g/ml,
respectively; and 0.998 g/ml endotoxin-free water. All solutions contained 0.1
mg/ml EDTA. The lipoprotein fractions VLDL, LDL, HDL2, and HDL3 were collected
by aspiration, dialyzed extensively against PBS at 4°C, and stored at –80°C
until use. Cholesterol concentrations in the isolated lipoprotein subfractions
were determined using an enzymatic colorimetric test from F. Hoffman LaRoche,
AG (Basel, Switzerland). The lipoprotein fractions used contained no detectable
LBP as assessed by LBP-specific ELISA (15).
Purification of LBP and production of LBP-depleted human serum.
LBP was isolated
from human plasma by
selective-affinity immunosorption as described previously (21). Plasma from healthy volunteers was kindly
provided by the local blood bank. In short, anti-hLBP mAb HM-14 was
cross-linked to CNBr-activated Sepharose (Pharmacia Biotech A/S) according to
the manufacturer’s instructions. Human plasma was applied to the anti-LBP
column, and unbound proteins were washed out with 0.5 M MgCl. Bound LBP was
eluted with 0.1 M Glycine-HCl buffer (pH 2.5). LBP-depleted serum was produced
by passing serum over the anti-LBP column. LBP-depleted serum contained less
LBP than 0.05% of normal serum as assessed by LBP-ELISA.
Preparation of
biotin-labeled LBP and LPS. And another 3 pages.
Discussion
The
structure and function of LBP are extensively studied; however, knowledge
concerning the in vivo forms or associations with other serum components and
the effect of these associations on the function of LBP is limited. In the
present study, we obtained evidence that LBP circulates in association with
apoB-containing lipoproteins in healthy persons and in septic patients. This
association is functional, as LBP bound to LDL and VLDL was observed to enhance
the LPS-binding capacity of these lipoproteins, a process known to result in
protection from the deleterious effects of LPS toxicity.
In search of
the factors involved in the association of LBP with apoB-containing
lipoproteins in serum, we found that this interaction is at least in part
mediated by an interaction of LBP with apoB. Although LBP was not associated
with apoA-I or HDL in serum, our experiments demonstrate that LBP does bind to
purified apoA-I. Evidence for a specific interaction of LBP with apoA-I is
supported by studies by Massamiri et al., who demonstrated that binding of LBP
to reconstituted HDL is partially blocked by antibodies against apoA-I (23). In the present study,
we observed a tenfold higher affinity of LBP for apoB compared with apoA-I
(Figure (Figure8c).8c). The amount of apoA-I molecules in serum, however, is
about 30 times higher than the amount of apoB under healthy conditions,
therefore, it is not possible to subscribe the predominant interaction of LBP
with LDL and VLDL to the higher affinity of LBP for apoB. It is possible that
LBP interacts with domains of apoA-I that are masked by incorporation in native
HDL. Furthermore, other constituents than apoB may contribute to the high
affinity of LBP for LDL and VLDL and consequently increase the competition for
binding of LBP to HDL.
The
LBP-transporting lipoproteins LDL and VLDL were also found to be the predominant
LPS-binding lipoproteins in normal human serum, an observation which is
supported by others (5, 24). Studies performed in
rodents report the preferential binding of LPS to HDL (4, 25–27). Relatively high HDL
levels in rodents seem to account for the predominant binding of LPS to HDL, as
high LDL levels in Watanabe heritable hyperlipidemic rabbits and high VLDL
levels in apoE knockout mice result in the predominant binding of LPS to LDL (5, 28) and to VLDL (J. Kuiper
personal communication), respectively. In contrast to rodents, the HDL:LDL/VLDL
ratio is low in humans, which most likely causes the differences observed for
the distribution of LPS among lipoproteins in humans and rodents.
Our data
further demonstrate that LBP associated with LDL and VLDL strongly enhances
binding of LPS to these lipoproteins in a dose-dependent fashion. However,
presence of LBP is not required for the interaction of LPS with LDL and VLDL.
ApoB, present in LDL and VLDL, was demonstrated to have high affinity for LPS
and therefore may account for LPS binding under LBP-free conditions. This
interaction of LPS with apoB is inhibited by presence of LBP, implicating
competition of LBP and LPS for apoB. It can be hypothesized from these data
that apoB functions as an anchor for LBP on LDL and VLDL. This bound LBP may
catalyze the binding of LPS to other lipoprotein components and thereby enhance
the total LPS-binding capacity. Given that others found that a lipid-lipid
interaction accounts for the association of LPS with lipoproteins (5) and have demonstrated
that LBP enhances binding of LPS to phospholipid membranes (29, 30), it is likely that LBP
linked to apoB enhances intercalation of LPS into the phospholipid membrane of
the lipoproteins. Other proteins present in LDL and VLDL may, however, also
contribute to the LPS-binding capacity of these lipoproteins. ApoE, present in
VLDL, for instance, was demonstrated to bind LPS (31). Most interestingly,
apoE-deficient mice display increased susceptibility to endotoxemia (32), the latter supporting
a possible role for apoE in the scavenging of LPS.
During infection, lipoproteins are proven to be fundamental
for the survival of the host (1, 2, 9, 10, 19). Under these
conditions, lipoprotein levels and
composition are known to be altered profoundly (33). These
changes prompted us to evaluate the interaction of
lipoproteins with LBP and endotoxins in septic serum. The changes in
lipoprotein composition and the more than tenfold enhanced LBP levels did not
significantly affect the distribution profile of LBP. In serum of septic
patients, LBP is also colocated predominantly with apoB (Figure (Figure10).10). These observations
indicate that during sepsis,
approximately tenfold more LBP is associated with LDL and VLDL. This is of
particular interest, as we observed that the LBP-induced upregulation of LPS
binding to these lipoproteins is dose-dependent. Only a small fraction of the
total LBP in septic sera migrates as free LBP. Whether there is a difference in
biologic activity between lipoprotein-associated and non–lipoprotein-associated
LBP in the host response to endotoxin is currently under our investigation. The
predominant incorporation of LPS in LDL and VLDL is also observed in septic
serum and does not seem to be influenced by the alterations in lipoprotein
composition and the presence of free LBP. Our results, which indicate that LPS
and LBP both predominantly bind to LDL and VLDL under septic conditions and
that apoB forms a binding site for LBP and LPS, are in line with the findings
of others (16) that in contrast
to apoA-I, apoB levels stay high during
infection. The remaining high concentration of apoB in LDL and VLDL seems
functional, as it may contribute to the enhanced binding of LBP and, as a
consequence LPS, to these lipoproteins during infection.
It is firmly established that binding of LPS to lipoproteins
reduces LPS toxicity (3). In accordance,
hypolipidemia results in enhanced
LPS-induced lethality in animals (2), and in humans,
low levels of cholesterol predict an
increased risk of death from infection (34), which emphasizes
the significance of lipoproteins in
protection from bacteria and their toxins. The protective function of
lipoproteins is considered to be due to an increase in the clearance of LPS by
formation of LPS-lipoprotein complexes and to prevention of its binding to
cells. In addition, it was recently demonstrated that lipoproteins, including
LDL, also promote the release of cell-associated LPS, which was proved to be
dependent on LBP (35). This inhibition
of endotoxin binding reduces activation of
monocytes and thereby the secretion of cytokines (3,35, 36). This protective
property of lipoproteins is not only
described for gram-negative bacteria, but also the toxic effects of fragments
of gram-positive bacteria are inhibited by LDL, a process catalyzed by the
presence of LBP (13). The beneficial
role for LDL in the host defense against
bacteria is supported by a study that demonstrates that LDL-receptor–deficient
mice with elevated circulating LDL concentrations are protected against lethal
endotoxemia and severe infections with gram-negative micro-organisms (19).
The uptake of
LPS into LDL, which we consider beneficial during acute infection, should be
considered as potentially harmful during chronic inflammation. Since we studied
LPS binding to LDL and VLDL, and not toxicity, we cannot exclude that LPS binds
to LDL and VLDL in a manner by which it retains some toxic activity. In the long
term, these complexes may play
a pathogenic role in the development of atherosclerosis. Although LDL protects
endothelial cells from acute LPS toxicity by formation of LPS-LDL complexes,
these complexes migrate across the endothelium and, via unknown pathways,
increase the secretion of monocyte chemotactic activity by endothelial cells (28). [In other words,
infections in the intima media of the artery walls and the LDL immune response contributes
to the formation of atheroma. Thus atheroma is like the formation of a boil
from the presences of bacteria, and the leakage of this boil like atheroma
results in ischemic events.} As a
consequence, transport of LDL-LPS complexes into the artery wall may initiate
an inflammatory response and provoke an atherosclerotic reaction.
In
conclusion, we found strong evidence for an association of LBP with
apoB-containing lipoproteins in the circulation of healthy persons. Also in
septic patients with extremely high circulating LBP concentrations, LBP is
predominantly bound to apoB-containing lipoproteins. Most interestingly, LBP
associated with LDL and VLDL enhances the LPS-binding capacity of these
lipoproteins in a dose-dependent manner. Accordingly, in serum from healthy
persons and from septic patients, LDL and VLDL are the predominant LPS-binding
lipoproteins. Overall, the data of this study suggest that LBP is a cofactor of
circulating LDL and VLDL, which facilitates the uptake of endotoxin by these
lipoproteins. This implies an important role for LBP/LDL and VLDL complexes in
the defense against bacteria and endotoxin.
Go to:
Acknowledgments
We thank M.
Poeze for the assistance in obtaining the serum of septic patients. This work
was supported by a grant of the Dutch Digestive Diseases Foundation, The
Netherlands References1. Read TE, et al. The protective
effect of serum lipoproteins against bacterial lipopolysaccharide. Eur Heart J. 1993;14:125–129. [PubMed]2. Feingold KR, et al. Role for
circulating lipoproteins in protection from endotoxin toxicity. Infect Immun. 1995;63:2041–2046. [PMC free article] [PubMed]3. Flegel WA, Wölpl A, Männel DN,
Northoff H. Inhibition of endotoxin-induced activation of human monocytes by human lipoproteins. Infect
Immun. 1989;57:2237–2245. [PMC free article] [PubMed]4. Ulevitch RJ, Johnston AR, Weinstein
DB. New function for high density lipoproteins. J Clin Invest. 1979;64:1516–1524.
[PMC free article] [PubMed]5. Van Lenten BJ, Fogelman AM, Haberland
ME, Edwards PA. The role of lipoproteins and receptor-mediated endocytosis in the transport of bacterial lipopolysaccharide.
Proc Natl Acad Sci USA. 1986;83:2704–2708.
[PMC free article] [PubMed]6. Victorov AV, et al. Composition
and structure of lipopolysaccharide-human plasma low density lipoprotein complex. Biochim
Biophys Acta. 1989;984:119–127. [PubMed]7. Netea MG, et al. Bacterial lipopolysaccharide
binds and stimulates cytokine producing cells before neutralization by endogenous lipoproteins can occur. Cytokine.
1998;10:766–772. [PubMed]8. Read TE, et al. Triglyceride-rich
lipoproteins improve survival when given after endotoxin in rats. Surgery. 1995;117:62–67. [PubMed]9. Harris HW, et al. Chylomicrons
alter the fate of endotxin, decreasing tumor necrosis factor release and preventing death. J
Clin Invest. 1993;91:1028–1034. [PMC free article] [PubMed]10. Harris HW, Grunfeld C, Feingold
KR, Rapp JH. Human very low density lipoproteins and chylomicrons can protect against endotoxin-induced death in mice. J Clin Invest. 1990;86:696–702. [PMC free article] [PubMed]11. Jack RS, et al. Lipopolysaccharide-binding
protein is required to combat a murine gram-negative bacterial infection. Nature. 1997;389:742–745. [PubMed]12. Wurfel MM, Kunitake ST, Lichenstein
H, Kane JP, Wright SD. Lipopolysaccharide (LPS)-binding protein is carried on lipoproteins and act as a cofactor in the neutralization
of LPS. J Exp Med. 1994;180:1025–1035. [PMC free article] [PubMed]13. Grunfeld C, et al. Lipoproteins
inhibit macrophage activation by lipoteichoic acid. J Lipid Res. 1999;40:245–252.
[PubMed]14. Lamping N, et al. LPS-binding
protein protects mice from septic shock caused by LPS or gram-negative bacteria. J Clin Invest.
1998;101:2065–2071. [PMC free article] [PubMed]15. Froon AHM, Dentener MA, Greve
JWM, Ramsay G, Buurman WA. Lipopolysaccharide toxicity regulating proteins in bacteremia. J
Infect Dis. 1995;171:1250–1257. [PubMed]16. Sammalkorpi K, Valtonen V, Kerttula
Y, Nikkilä E, Taskinen M-R. Changes in serum lipoprotein pattern induced by acute infections. Metabolism.
1988;37:859–865. [PubMed]17. Alvarez C, Ramos A. Lipids,
lipoproteins and apoproteins in serum during infection. Clin Chem. 1986;32:142–145.
[PubMed]18. Cabana VG, Siegel JN, Sabesin
SM. Effects of the acute phase response on the concentration and density distribution of plasma lipids and apolipoproteins.
J Lipid Res. 1989;30:39–49. [PubMed]19. Netea MG, et al. Low-density
lipoprotein receptor deficient mice are protected against lethal endotoxemia and severe gram-negative infections. J Clin Invest. 1996;97:1366–1372. [PMC free article] [PubMed]20. Terpstra AHM, Woodward CJH,
Sanchez-Muniz FJ. Improved techniques for the separation of serum lipoproteins by density gradient ultracentrifugation: visualization
by prestaining and rapid separation of serum lipoproteins from small volumes of serum. Anal
Biochem. 1981;111:149–157. [PubMed]21. Vreugdenhil ACE, et al. Lipopolysaccharide
binding protein and serum amyloid A secretion by human intestinal epithelial cells during the acute phase response. J Immunol. 1999;163:2792–2798. [PubMed]22. Kunitake ST, et al. Identification
of proteins associated with apolipoprotein A-I-containing lipoproteins purified by selected-affinity immunosorption. Biochemistry. 1994;33:1988–1993. [PubMed]23. Massamiri T, Tobias PS, Curtiss
LK. Structural determinants for the interaction of lipopolysaccharide binding protein with purified high density lipoproteins:
role of apolipoprotein A-I. J Lipid Res. 1997;38:516–525.
[PubMed]24. Eggesbo JB, Lyberg T, Aspelin
T, Hjermann I, Kierulf P. Different binding of 125I-LPS to plasma proteins from persons with high or low HDL. Scand J Clin Lab Invest. 1996;56:533–543. [PubMed]25. Tobias PS, Ulevitch RJ. Control
of lipopolysaccharide-high density lipoprotein binding by acute phase protein(s) J Immunol.
1983;131:1913–1916. [PubMed]26. Munford RS, Andersen JM, Dietschy
JM. Sites of tissue binding and uptake in vivo of bacterial lipopolysaccharide-high density lipoprotein complexes. J Clin Invest. 1981;68:1503–1513. [PMC free article] [PubMed]27. Munford RS, Hall CL, Dietschy
JM. Binding of Salmonella typhimurium lipopolysaccharides to rat high-density lipoproteins. Infect
Immun. 1981;34:835–843. [PMC free article] [PubMed]28. Navab M, Hough GP, van Lenten
BJ, Berliner JA, Fogelman AM. Low density lipoproteins transfer bacterial lipopolysaccharides across endothelial monolayers
in a biologically active form. J Clin Invest. 1988;81:601–605.
[PMC free article] [PubMed]29. Schromm AB, et al. Lipopolysaccharide-binding
protein mediates CD14-independent intercalation of lipopolysaccharide into phospholipid membranes. FEBS
Lett. 1996;339:267–271. [PubMed]30. Wurfel MM, Wright SD. Lipopolysaccharide-binding
protein and soluble CD14 transfer lipopolysaccharide to phospholipid bilayers. J Immunol.
1997;158:3925–3934. [PubMed]31. Rensen PCN, et al. Human recombinant
apolipoprotein E redirects lipopolysaccharide from Kupffer cells to liver parenchymal cells in rats in vivo. J
Clin Invest. 1997;99:2438–2445. [PMC free article] [PubMed]32. De Bont N, et al. Apolipoprotein
E knock-out mice are highly susceptible to endotoxemia and Klebsiella pneumoniae infection. J
Lipid Res. 1999;40:680–685. [PubMed]33. Auerbach BJ, Parks JS. Lipoprotein
abnormalities associated with lipopolysaccharide-induced lecithin: cholesterol acyltransferase and lipase deficiency. J Biol Chem. 1989;264:10264–10270. [PubMed]34. Jacobs D, et al. Report of
the conference on low blood cholesterol: mortality associations. Circulation. 1992;86:1046–1060. [PubMed]35. Kitchens RL, Wolfbauer G, Albers
JJ, Munford RS. Plasma lipoproteins promote the release of bacterial lipopolysaccharide from the monocyte cell surface. J Biol Chem. 1999;274:34116–34122. [PubMed]36. Cavaillon J-M, Fitting C, Haeffner-Cavaillon
N, Kirsch SJ, Warren HS. Cytokine response by monocytes and macrophages to free and lipoprotein-bound lipopolysaccharide.
Infect Immun. 1990;58:2375–2382. [PMC free article] [PubMed]
|