An
article which proves the case that infectious agents are the cause of
cardiovascular disease and all its consequences. For more
journal evidence go to http://healthfully.org/rl/id8.html,
more by Prof. Uffe Ravnskov at id5/html
on the Cholesterol myth—jk.
http://www.reseachchgate.net/publication/223976877_Infections..._Pathogenesis_of_atherosclerosis/file
/ e0b49527cbf19904d3.pdf
REVIEW
ARTICLE:
The American
Journal of the Medical Sciences _ Volume 0, Number
0, Month 2012
Infections May be Causal in the Pathogenesis
of Atherosclerosis
Uffe Ravnskov, MD, PhD and
Kilmer S. McCully, MD
Abstract: There
is a universal lack of exposure response between degree of lipid lowering and
the outcome in clinical and angiographic trials questioning the current view on
atherogenesis. However, there are numerous
observations and experiments suggesting that microorganisms may play a causal
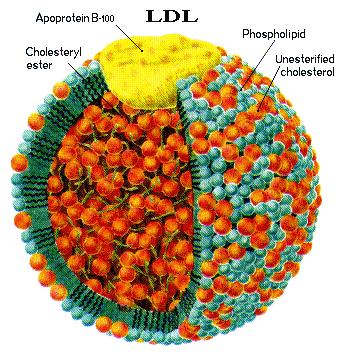
role. A clue is the fact that the lipoproteins
constitute an innate immune system by binding and inactivating microorganisms
and their toxic products through formation of circulating complexes. Their
size may increase in the presence of hyperhomocysteinemia because homocysteine
reacts with low-density lipoprotein (LDL) to form homocysteinylated LDL
aggregates. Autoantibodies against homocysteinylated or oxidized LDL may also enhance
the aggregation. Because of the high extra-capillary pressure, such aggregates
may obstruct arterial vasa vasorum [arteriola and that provide oxygen to cells
within the artery’s walls] producing ischemia and cell death within the
arterial wall leading to the creation of
a vulnerable plaque. The many epidemiological observations, clinical findings
and laboratory experiments that conflict
with the cholesterol hypothesis are in good accordance with ours.
Key Indexing Terms: Atherosclerosis;
Cholesterol; Lipoproteins; Hypothesis; Infections; Microorganisms; Vulnerable
plaque; Homocysteine. [Am J. Med Sci 2012;0(0):1–4.]
CONTRADICTIONS TO THE CURRENT VIEW
According
to the cholesterol hypothesis, atherosclerosis is initiated by endothelial
dysfunction caused by hypercholesterolemia, hyperhomocysteinemia or other toxic
factors. Endothelial dysfunction is said to allow LDL-cholesterol and monocytes
to enter the arterial wall, where LDL-cholesterol becomes oxidized and taken up
by the macrophages. These events are considered to cause inflammation
and to be the starting point of atherosclerosis. There are obvious contradictions
to this interpretation:
1. There is
no association between the concentration of LDL cholesterol in the blood and
the degree of endothelial dysfunction.1
2. The atherosclerotic
plaques seen in extreme hyperhomocysteinemia caused by inborn errors of
methionine metabolism do not contain any lipids despite pronounced endothelial
damage.2
3. No autopsy
study of unselected, adult individuals has found an association between serum
cholesterol and the degree of atherosclerosis.3 Moreover,
there is no association between serum cholesterol and the degree of coronary calcification
measured by electron beam tomography.4 To quote
the authors: “There
were no
significant differences
in the calcium
scores throughout the entire range of all lipid parameters; calcium percentiles
were virtually identical within lipid value subgroups.”
4. High
cholesterol is not a risk factor for coronary heart disease in women or in old
individuals. In fact, more than a dozen studies have found that old people with
high cholesterol live the longest.5–8
5. According
to the 30-year follow-up study from Framingham, both heart and total mortality
were the highest among those whose cholesterol had decreased.9
6. Trials
with anti-inflammatory
drugs have demonstrated an increase of cardiovascular death in the treatment
groups questioning that inflammation is
the inciting cause.10,11
According
to Karl Popper, the idea that high cholesterol causes atherosclerosis is a true
scientific
hypothesis
because it is falsifiable. As the cited contradictions have
effectively falsified the cholesterol hypothesis,
there are reasons to consider other ideas.
In a
previous article, we presented a new hypothesis. suggesting a crucial role of
infections.12 Since
its
publication, we have identified more
studies in the past which are in support, and several new studies have been
published presenting data that are in accordance with our hypothesis.
CARDIOVASCULAR
DISEASE IS ASSOCIATED WITH INFECTION
Most
investigators consider the association between infections and cardiovascular
disease as a secondary phenomenon, but by several reasons it may not be that
simple. Cardiovascular mortality increases during influenza
epidemics, and about a third of patients with acute cardiovascular disease have
had an infection immediately before onset.12 Bacteremia
and periodontal infections are associated with an increased risk of
cardiovascular disease,12 and
Piconi et
al13 found that treatment
of periodontal
infections improved endothelial function and reduced the intima-media
thickening of the carotid arteries to a much higher degree than seen in any
cholesterollowering trial. Furthermore, serological markers of infection are increased
in patients with cardiovascular disease,6 and
bacteremia and sepsis are found frequently in patients with cardiogenic shock
due to myocardial infarction.12
One
hundred years ago, bacteria and viruses were considered as the main cause of atherosclerosis.
The main arguments were the high frequency of arterial lesions in patients who
died from typhoid fever and the high prevalence of arteriosclerotic radial
arteries in those who survived,12 and
the association
between the degree of atherosclerosis in people who had died from an infectious
disease and the duration of the preceding infection. Said by Klotz and Manning:
“There is every
indication that the
production of tissue in the intima is the result of a direct irritation of that
tissue by the presence of infection or toxins,”12 and
William Osler described the vulnerable plaque as an atherosclerotic pustule.12
______________________________________________________________
From the Magle Stora Kyrkogata
9 (UR), Lund, Sweden; Pathology and
Laboratory Medicine Service
(KSM), VA Boston Healthcare System,
West Roxbury,
Massachusetts; and Department of Pathology (KSM),
Harvard Medical School, Boston, Massachusetts.
Submitted October 19, 2011; accepted
in revised form January 13, 2012.
K.S. McCully holds U.S. patents
on
synthetic homocysteine thiolactone derivatives for use in therapy of
degenerative diseases of aging.
Correspondence: Uffe Ravnskov,
MD,
PhD, Magle Stora Kyrkogata 9, 22350 Lund, Sweden (E-mail: ravnskov@tele2.se).
Page 1
The
classical study of early atherosclerosis in young American soldiers killed in
Korea is frequently cited as proof that atherosclerosis starts in early
adulthood.14 In
that
study, 77.3% had gross evidence of coronary disease and 15% had more than 50%
luminal narrowing. However, such severe changes have never been observed in
autopsy studies of young people who have died from other causes. The
explanation may be that many of these soldiers had severe, infected wounds before
they died. As the author stated, “thrombosis
occurred especially in cases in which extensive trauma and shock exerted their
influence.” In a
postmortem study of several thousand victims in the concentration camp in
Dachau, Blaha15 found
extensive atherosclerosis in individuals younger than 35 years. Many had severe
infections, and the degree of arteriosclerosis was related to the duration of
internment in the camp. Other than severe stress, there was no dietary
cholesterol or saturated fat, no smoking, no lack of exercise, no obesity or
other risk factors for arteriosclerosis. Much evidence indicates that
atherosclerosis starts in childhood and is associated with infectious diseases.
Liuba et al16 studied
infected children by high-resolution ultrasound and found narrowing of the
coronary arteries in those who died and thickening of the carotid intima-media
layer in those who survived.
EXPERIMENTAL EVIDENCE
Fabricant
et al17 induced visible
atherosclerotic changes in
chickens by infecting them with Marek’s
disease herpes virus, and Damy et al18 worsened
atherosclerosis in hypercholesterolemic mice by Chlamydia
pneumoniae or Mycoplasma
pneumoniae. Recently, Birck et al19 produced
signs of early atherosclerosis in normo- and hypercholesterolemic minipigs by
infectin g them with C
pneumoniae, alone or together with influenza
virus. Vascular damage and endothelial dysfunction were most prominent in the
coinfected animals but less pronounced in the hypercholesterolemic than in the
normocholesterolemic pig, also a contradiction to the current view, but in accordance
with our idea that the lipoproteins may be protective due to their
antimicrobial properties.
If
atherosclerosis is caused by microorganisms, vaccination or antibiotics should
be able to prevent cardiovascular disease. Some randomized controlled trials
have indeed shown benefit, either
from influenza
vaccination or from short-term antibiotic treatment, but just as many have
failed. These results are not contradictory, because Ott et al20 have
identified remnants of
more than 50
bacterial species within atherosclerotic plaques and other investigators have
found various virusspecies as well.21 It
is
unlikely that a single antibiotic used during a few weeks should be able to
eliminate more than 50 different bacterial or viral species.
ROLE OF THE LIPOPROTEIN IMMUNE SYSTEM
Despite
many associations between infections and cardiovascular disease, little
attention has been paid to the lipoproteins as mediators of the immune system.
In 1939, Todd, Coburn and Bradford Hill found that a serum factor named antistreptolysin-S
was not an antibody as previously thought, because its titre fell in rheumatic
fever at the peak of the clinical symptoms and during convalescence.12
Ten
years later, Humphrey located antistreptolysin-S within the lipoprotein
fraction of the blood. Since then, a dozen research groups have documented that
antistreptolysin-S is identical with the lipoproteins and constitutes a
nonspecific
host
defense system that is able to bind and neutralize not only streptolysin-S but
also other endotoxins and a large number of bacterial and viral species.12
In
vitro studies have shown that human LDL inactivates up to 90% of Staphylococcus
aureus a-toxin
and an
even larger fraction of bacterial lipopolysaccharide (LPS). Compared with normal
rats, hypocholesterolemic rats injected with LPS have a markedly increased
mortality, which can be ameliorated by injecting purified
human LDL, and hypercholesterolemic mice challenged with LPS or live bacteria
have an 8-fold higher LD50 compared with normal rats.12
Studies
on human beings are in support as well. For instance, a review of 19 cohort
studies found that low serum cholesterol is a risk factor for infectious
diseases.22 It
has been argued
that low cholesterol is a secondary phenomenon. However, in a 15-year follow-up
study of more than 100,000 healthy people those with low cholesterol had been
admitted significantly
more
often to hospital because of an infectious disease.22 Obviously,
the low cholesterol could not be secondary to a disease that they had not yet
developed. Also in agreement is that before 1900, when infectious diseases were
the commonest cause of death, the lifespan of people with familial
hypercholesterolemia was longer than that of the general population.23
Infectious
diseases cause dyslipidemia,24,25 but
to call the lipid pattern atherogenic may be misleading. The altered lipid profile
may instead reflect
the body’s
response to infections. A recent report by Pletcher et al26 showed
that time-averaged cumulative nonoptimal lipid levels in young adults were
associated with an increased coronary calcium score later in life. The periodic
dyslipidemia was interpreted as
the first sign of atherosclerosis.
As no
previous study of unselected individuals has shown an association between the
blood lipids and degree of atherosclerosis,3,4 a
more likely interpretation is that the increased calcium score may have been
the result of spontaneously resolved infections and that the calcified
lesions later in life may be scars after healed infections.
CREATION OF THE VULNERABLE PLAQUE
We
suggest that in the case of chronic or severe acute infections, arterial vasa
vasorum may be obstructed by complexes formed between lipoproteins,
microorganisms and their toxic products. Their size may increase in the
presence of antibodies against oxidized or homocysteinylated LDL. Homocysteine reacts
with LDL, and homocysteinylated LDL aggregates are phagocytozed by macrophages
to form foam cells.27 In addition,
hyperhomocysteinemia causes endothelial dysfunction, narrowing the lumen of
capillaries and leading to trapping of lipoprotein aggregates within vasa
vasorum in areas of high tissue pressure.28 Because
vasa vasorum are end arteries, their blockage by this process may cause
ischemic cell death of the arterial wall and lead to the creation of a
vulnerable plaque.
It is
generally accepted that rupture of a vulnerable plaque is the main cause of
most arterial thromboses. If the vulnerable plaque is a pustule, as suggested
by Osler, its temperature should be higher than its surroundings, which was found
to be the case.12 Moreover,
the
symptoms and the laboratory findings in
acute myocardial infarction are similar to those of an infectious disease.
PATHOLOGICAL EVIDENCE
If
the starting point of atherosclerosis is in the vasa vasorum, inflammation
should be most pronounced in the adventitia. In accordance, Higuchi et al29,30
found medial thinning and 4 times more lymphocytes and monocytes
and more microvessels in the adventitia beneath vulnerable plaques than beneath
stable ones, and in the vasa vasorum, monocytes containing
elementary bodies of C pneumoniae
were
seen (Figure 1). (USE LINK FIGURE—couldn’t copy, notes on at end of article.
Maiellaro
and Taylor31 have
also
presented arguments for an “outside-in” mechanism
that may work in concert with the conventional “inside-out” one.
They point out the rich presence in the adventitia of macrophages and T and B
lymphocytes and suggest that the latter may generate antibodies against inflammatory
antigens. The nature of these antigens is still undetermined; the authors
suggest that heat shock proteins, modified
lipoproteins and other surface antigens may be responsible. We suggest that
microbes, or microbial antigens, released from the lipoprotein complexes in
case of tissue anoxia caused by the obstruction of vasa vasorum are causing the
inflammatory response.
In accordance,
Nicolaou et al32 have
shown
that 9 bacteria, representing those most frequently reported to be present in
human atheroma induced, foam cell formation of monocytes and macrophages.
If
the vulnerable plaque is a vascular pustule, it should contain infectious
agents, and this is also the case. Using electron microscopy and in
situ hybridization, Higuchi et al30,33
have
detected M pneumoniae, C
pneumoniae and also archaeal bodies situated in the
lipid core of ruptured vulnerable plaques. These microorganisms attract mainly
lymphocytes, but because neutrophilic granulocytes are frequently found in and
around the vulnerable plaques,6 other
microorganisms may play a role as well, an issue for future research.
CONCLUSIONS
The
function of lipoproteins in the immune system has been ignored in the
literature about lipids and atherosclerosis, although it may provide the key to
understanding the pathogenesis of atherosclerosis. We suggest that aggregates
formed between the lipoproteins and microbes and enlarged by antibodies against
oxidized or homocysteinylated LDL may obstruct arterial vasa vasorum because of
the high extracapillary tissue pressure. By this process, the arterial wall may
become anoxic, leading to an accumulation of toxic substances and microorganisms
in the arterial wall inciting the inflammatory
response. The vulnerable plaque may simply be a microabscess, as first
suggested by William Osler 100 years ago.
ACKNOWLEDGMENT
We are indepted to Professor Maria de Lourdes Higuchi for having
supplied the figure.
REFERENCES
1. Reis
SE, Holubkov R, Conrad Smith AJ, et al; WISE
Investigators. Coronary
microvascular dysfunction is highly prevalent in women with chest pain in the
absence of coronary artery disease: results from the NHLBI WISE study. Am Heart
J 2001;141:735–41.
2. McCully
KS. Vascular
pathology of homocysteinemia: implications for the pathogenesis of
arteriosclerosis. Am J Pathol 1969;56:111–28.
3. Ravnskov
U. Is
atherosclerosis caused by high cholesterol? Q J Med 2002;95:397–403.
4. Hecht
HS, Superko HR, Smith LK, et al. Relation of coronary artery calcium identified by electron
beam tomography to serum lipoprotein levels and
implications for treatment. Am J Cardiol 2001;87:406–12.
5. Schatz
IJ, Masaki K, Yano K, et al. Cholesterol
and all-cause mortality in elderly people from the Honolulu Heart Program: a
cohort
study. Lancet 2001;358:351–5.
6. Onder
G, Landi F, Volpato S, et al. Serum
cholesterol levels and in-hospital mortality in the elderly. Am J Med
2003;115:265–71.
7. Ulmer
H, Kelleher C, Diem G, et al. Why
Eve is not Adam: prospective follow-up in 149650 women and men of cholesterol
and other
risk factors related to cardiovascular and all-cause mortality.
J Womens Health (Larchmt) 2004;13:41–53.
8. Schupf
N, Costa R, Luchsinger J, et al. Relationship between plasma lipids and all-cause mortality in
nondemented elderly. J Am Geriatr Soc 2005;53:219–26.
9. Anderson
KM, CastelliWP, Levy D. Cholesterol
and mortality. 30 years of follow-up from the Framingham study. JAMA
1987;257:2176–80.
10. Johnsen
SP, Larsson H, Tarone RE, et al. Risk of hospitalization for myocardial infarction among users of
rofecoxib, celecoxib, and other NSAIDs: a population-based case-control study.
Arch Intern Med 2005; 165:978–84.
11. Hippisley-Cox
J, Coupland C. Risk of
myocardial infarction in patients taking cyclo-oxygenase-2 inhibitors or
conventional non-steroidal anti-inflammatory
drugs: population based nested case-control analysis. BMJ 2005;330:1366–72. _ 2012 Lippincott Williams &
Wilkins
12. Ravnskov
U, McCully KS. Vulnerable
plaque formation from obstruction of Vasa vasorum by homocysteinylated and
oxidized lipoprotein aggregates complexed with microbial remnants and LDL
autoantibodies. Ann Clin Lab Sci 2009;39:3–16.
13. Piconi
S, Trabattoni D, Luraghi C, et al. Treatment of periodontal disease results in improvements in
endothelial dysfunction and reduction of the carotid intima-media thickness.
FASEB J 2009;23:1196–204.
14. Enos
WF, Holmes RH, Beyer J. Coronary
disease among United States soldiers killed in action in Korea; preliminary
report. JAMA
1953;152:1090–3.
15. Blaha
F. [Observations
on the pathogenesis of arteriosclerosis from an experience in a concentration
camp.] _Cas Lék _Ces [J Czech Physicians] 1958;97:86–9.
16. Liuba
P, Persson J, Luoma J, et al. Acute
infections in children are accompanied by oxidative modification of LDL and decrease of HDL cholesterol,
and are followed
by thickening of carotid intima-media. Eur Heart J 2003;24:515–21.17. Fabricant
CG, Fabricant J, Litrenta MM, et al. Virus-induced atherosclerosis. J Exp Med 1978;148:335–40.
18. Damy
SB, Higuchi ML, Timenetsky J, et al. Mycoplasma pneumonia and/or Chlamydophila
pneumoniae inoculation
causing different aggravations in cholesterol-induced atherosclerosis in apoE
KO male mice. BMC Microbiol 2009;9:194–201.
19. Birck
MM, Pesonen E, Odermarsky M, et al. Infection-induced coronary dysfunction and systemic inflammation
in piglets are dampened in hypercholesterolemic milieu.
Am J Physiol Heart Circ Physiol 2011; 300:H1595–601.
20. Ott
SJ, El Mokhtari NE, Musfeldt M, et al. Detection of diverse bacterial signatures in atherosclerotic
lesions of patients with coronary heart disease. Circulation 2006;113:929–37.
21. Shi
Y, Tokunaga O. Chlamydia
pneumoniae and multiple
infections in the aorta contribute to atherosclerosis. Pathol Int 2002;52:755–63.
22. Ravnskov
U. High
cholesterol may protect against infections and atherosclerosis.Q J Med
2003;96:927–34.
23. Sijbrands
EJ, Westendorp RG, Defesche JC, et al. Mortality over two centuries in large pedigree with familial hypercholesterolaemia:
family tree mortality study. BMJ 2001;322:1019–23.
24. Apostolou
F, Gazi IF, Kostoula A, et al. Persistence
of an atherogenic lipid profile after treatment of acute
infection with Brucella. J Lipid Res 2009;50:2532–9.
25. Gidding
SS, Stone NJ, Bookstein LC, et al. Month-to-month variability of lipids, lipoproteins, and
apolipoproteins and the impact of acute infection in adolescents. J Pediatr
1998;133:242–6.
26. Pletcher
MJ, Bibbins-Domingo K, Liu K, et al. Nonoptimal lipids commonly present in young adults and coronary
calcium later in life: the CARDIA (Coronary Artery Risk Development in Young
Adults) study. Ann Intern Med 2010;153:137–46.
27. Naruszewicz
M, Mirkiewicz E, Olszewski AJ, et al. Thiolation of low-density lipoprotein by homocysteine
thiolactone causes increased aggregation and altered interaction with cultured
macrophages. Nutr Metab Cardiovasc Dis 1994;4:70–7.
28. McCully
KS. Chemical
pathology of homocysteine. IV. Excitotoxicity, oxidative stress, endothelial
dysfunction, and inflammation. Ann Clin Lab Sci
2009;3:219–32.
29. Higuchi
ML, Gutierrez PS, Bezerra HG, et al. Comparison between adventitial and intimal inflammation
of ruptured and nonruptured atherosclerotic plaques in
human coronary arteries. Arq Bras Cardiol 2002; 79:20–4.
30. Higuchi
ML, Sambiase N, Palomino S, et al. Detection of Mycoplasma
pneumoniae and Chlamydia
pneumoniae in ruptured atherosclerotic plaques.
Braz J Med Biol Res 2000;33:1023–6.
31. Maiellaro
K, Taylor WR. The role of
the adventitia in vascular inflammation. Cardiovasc Res
2007;75:640–8.32. Nicolaou G, Goodall AH,
Erridge C. Diverse
bacteria promote macrophage foam cell formation via toll-like
receptor-dependent lipid body biosynthesis. J atheroscler Thromb 2012;19:137–48.
33. Higuchi
ML, Santos MH, Roggério A, et al. A role for archaeal organisms in development of atherosclerotic
vulnerable plaques and myxoid matrices. Clinics (Sao Paulo) 2006;61:473–8. Ravnskov
and McCull
The information
as part of journal images (not
available for copy because of the internet formation of pdf document. FIGURE 1.
Microscopic evidence. (A)
Light microscopy of a ruptured vulnerable plaque demonstrates adventitial inflammation,
injured media and fragmented internal and external elastic membranes. Note that
the inflammation
is most pronounced in the adventitia. Movat; scale bar: 1 mm.
(B)
Electron microscopy of a capillary in the adventitia demonstrates a monocyte
containing cytoplasmic elementary bodies of Chlamydia pneumoniae, characterized by the
typical pear shape because of expansion of the external membrane. Araldite and
OsO4; scale bar: 0.5 mm. (C) Light
microscopy of semithin section of a block before electron microscopy analysis demonstrates
adventitial capillaries containing many monocytes and surrounding macrophages.
Electron microscopy demonstrates cytoplasmic elementary bodies of C pneumoniae. Araldite and
Toluidine Blue: scale bar: 10 mm.
Atherosclerosis
and Infections