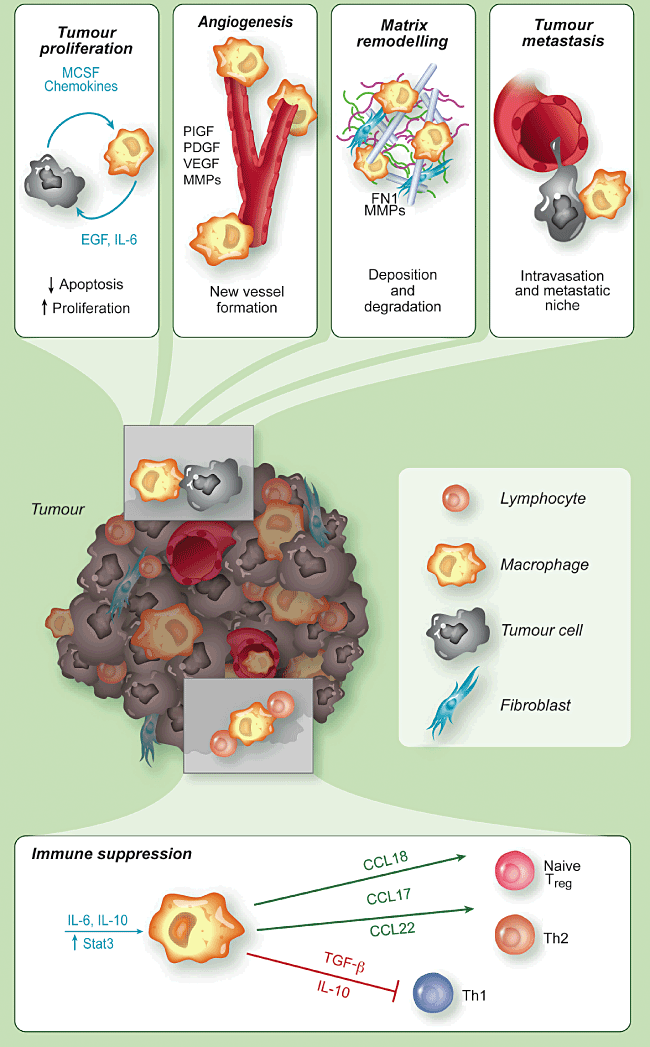
Figure 2.
Pro-tumour
functions of tumour-associated macrophages (TAM). TAM promote the survival of
neoplastic cells from apoptotic stimuli and their proliferation, by producing
several growth factors and cytokines [e.g. epithelial growth factor (EGF),
interleukin (IL)-6], and the tumour angiogenesis, via vascular endothelial
growth factor (VEGF), matrix metalloproteinases (MMPs) and other angiogenic
factors. TAM have an intense proteolityic activity and degrade the
extracellular matrix, but also produce matrix proteins, such as fibronectin
(FN1). They favour tumour cell intravasation and dissemination to distant
sites. TAM have immune suppressive functions by producing IL-10 and
transforming growth factor (TGF-β) which suppress T helper type 1 (Th1)
lymphocytes, and by secreting chemokines (e.g. CCL17, CCL18, CCL22) which
recruit lymphoid cells devoid of cytotoxic activity (Th2, naive lymphocytes) or
having suppressive functions [regulatory T cells (Treg)].
TAM
are key effectors of
the ‘angiogenic switch’ where the balance between pro- and anti-angiogenic
factors, commonly present in tissues, tilts towards a pro-angiogenic outcome [84–87]. In hypoxic conditions the transcription factor HIF-1alpha
induces in TAM the production of VEGF and the angiogenic chemokine CXCL8 [88].
In
addition, a unique
subset of monocytes has been identified recently in tumours by the expression
of the angiopoietin (Ang) receptor Tie-2, named Tie-2 expressing monocytes
(TEMs) [89] Thus, TAM and related myeloid
cells directly produce angiogenic
factors and are also a major source of proteolytic enzymes which mobilize VEGF
from extracellular matrix stores, indirectly sustaining tumour angiogenesis [90].
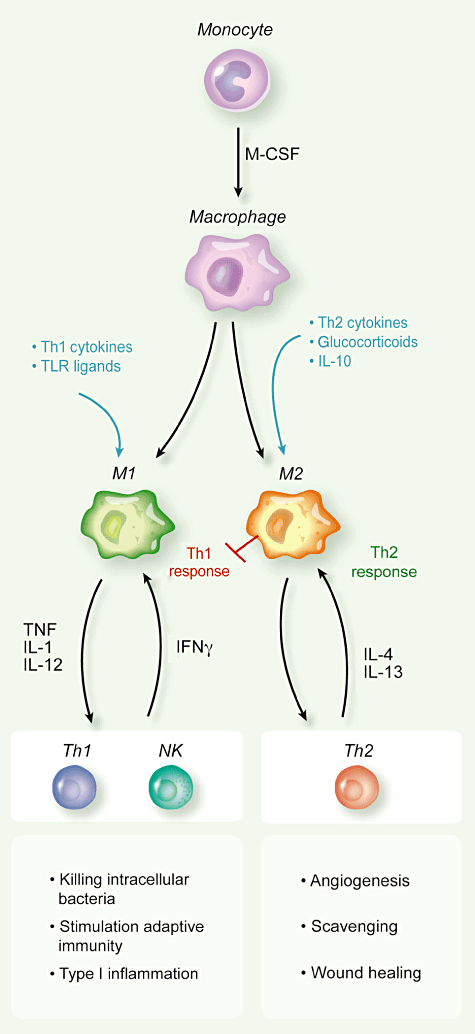
It is emerging
that the
expression pattern of genes that regulate iron homeostasis is distinct in
polarized macrophages. For instance, M1 macrophages showed ferroportin repression and H ferritin induction, thus
favouring iron sequestration, whereas M2 macrophages had an inverse expression
profile (ferroportin up-regulation and down-regulation of H ferritin and
haem oxygenase) and enhanced iron release [91]. Therefore, M1 cells mediate iron retention to control pathogen
expansion during the acute phase of inflammation, while M2 cells donate iron
that is important to tissue repair in the resolution phase. Interestingly, haem
oxygenase 1 is highly expressed in TAM; it is tempting to speculate that the increased
iron availability in the
tumour microenvironment might represent a previously unknown mechanism that
underlies the tumour-promoting activity of TAM [92].
TAM are probably the most active contributors to the incessant
matrix remodelling present within tumours, as they produce several MMPs and
other proteolytic enzymes [93]. Tumour cells exploit the ECM degradation mediated by TAM to invade
locally, penetrate into vessels and disseminate to give distant metastasis [94]. TAM aiding cancer cell
invasion have been visualized directly in experimental tumours in
vivo by
multi-photon
microscopy: by using fluorescently labelled cells, Wyckoff and colleagues
showed that tumour cell intravasation occurs next to perivascular macrophages
in mammary tumours [94,95]. Further, it has been shown recently that cathepsin protease
activity, by IL-4-stimulated TAM, promotes tumour invasion [96]. IL-4 is produced by tumour-infiltrating CD4 T cells and there
is mounting evidence of its relevance in the polarization of macrophages with
pro-tumour functions [14,16]. The chemokine CCL18
produced by TAM has been shown recently to play a critical role in promoting
breast cancer invasiveness by activating tumour cell adherence to ECM [97].
We
found recently that
human TAM and in-vitro tumour-conditioned
macrophages express high levels of the migration stimulation factor (MSF) [40], a truncated isoform of fibronectin [98]. Macrophage-secreted MSF displays potent chemotactic activity
to tumour cells in vitro [40], confirming that the pro-invasive phenotype of cancer cells is
modulated by macrophage products released in the tumour microenvironment.
Further support
to the
concept of a reciprocal interaction between tumour cells and TAM was provided
by a recent paper where SNAIL-expressing keratinocytes became locally invasive
after macrophage recruitment elicited by M-CSF [99].
Tumour macrophages have the ability to suppress the adaptive immune
response, thus contributing directly to the phenomenon of immune evasion of
cancer [1]. TAM are poor antigen-presenting cells, have defective IL-12
secretion [100], produce IL-10 and TGF-β and inhibit T cell proliferation [27,36,101]. At least some of these immune-suppressive activities of TAM
are mediated by over-activation of the transcription factor STAT3. In immune
cells STAT3 enables suppression of tumour immunity by opposing STAT1-regulated
Th1 anti-tumour immune responses and promoting the differentiation of immature
myeloid cells with suppressive activity [102].
Myeloid-derived
suppressor cells (MDSC), identified in tissues and lymphoid organs of
tumour-bearing hosts, contribute to tumour-induced immune suppression [101,103–105]. These cells share properties and gene expression profiles with
M2-polarized TAM, yet also display distinct features [106]. MDSC use two enzymes involved in the arginine metabolism to
control T cell response: inducible nitric oxide synthase (NOS2) and arginase
(Arg1), which deplete the milieu of arginine, causing peroxinitrite generation
and T cell apoptosis [107].
The
immune-suppressive
activity of TAM is also exerted indirectly by their release of chemokines (e.g.
CCL17 and CCL22) that preferentially attract Th1, Th2 lymphocytes and
regulatory T cells (Treg), devoid of cytotoxic
functions [72]. The chemokine CCL18, produced abundantly by TAM from human
ovarian carcinoma [108], recruits naive T cells, which eventually turn into anergic
cells within a microenvironment dominated by M2 macrophages and immature DC [109,110].
In
line with the above
experimental evidence, in the majority
of human tumours high numbers of infiltrating TAM have been associated
significantly with advanced tumours and poor patient prognosis [11,15,42,111]. There are, however, notable exceptions to this pro-tumour
phenotype, probably dictated by TAM functional polarization. One such exception is
human colorectal
cancer, where some studies have reported that TAM density is associated with
better prognosis [112–114]. The localization of TAM within colorectal cancers appears to
be of primary importance: the number of peritumoural macrophages with high
expression of co-stimulatory molecules (CD80 and CD86), but not of those within
the cancer stroma, was associated with improved disease-free survival [115,116].
Specific
TAM subsets
identified by surface markers may have predictive values: in lung
adenocarcinoma, the number of TAM CD204+ (scavenger receptor) showed a strong association with poor
outcome, while the total CD68+ population did not [117].
Macrophage-related
gene
signatures have been identified in human tumours such as ovarian and breast
cancer, soft tissue sarcoma and follicular B lymphoma [118–121]; in classic Hodgkin's lymphoma, tumours with increased number
of CD68+TAM were associated significantly with shortened
progression-free survival [122].
In
recent years there has
been increasing evidence that TAM and related myeloid cells with pro-angiogenic
(Tie-2+monocytes) and/or immune suppressive functions
(MDSC) [101,103,104,123] are implicated strongly in
the failure of anti-tumour therapies [124,125]. Accumulation of myeloid CD11b+Gr1+ cells (including TAM,
MDSC and immature cells) in tumours renders them refractory to angiogenic
blockade by VEGF antibodies [126]. This effect was traced to a VEGF-independent pathway driven by
the granulocyte colony-stimulating factor (G-CSF)-induced protein Bv8 [127]. Further, depletion or pharmacological inhibition of TEMs in
tumour-bearing mice markedly increased the efficacy of therapeutic treatment
with a vascular-disrupting agent. Overall, these data indicate that myeloid
cells, including TAM, considerably limit the clinical efficacy of
anti-angiogenic therapies [124].
Targeting of
TAM in tumours
The
pro-tumour functions
of TAM make these cells attractive targets of biological anti-cancer therapies.
Macrophage depletion in experimental
settings has been successful to limit tumour growth and metastatic spread [25,128,129], and to achieve better responses to conventional chemotherapy
and anti-angiogenic therapy [101,103,123–125]. [Unfortunate this is improve
not cure, because once the TAM
relationship has been established and cancer cells have acquired microphage
invasive properties, stopping that association might not reverse it. To increase
by adding another drug to the
cocktail offered a cancer patient thus far has NOT resulted in a cure, at best
it extends remission a few months at the cost dollars and side effects--jk]
A number of
studies have shown that the bisphosphonate clodronate encapsulated in
liposomes is an
efficient reagent for the depletion of macrophages in vivo. Clodronate-depletion of
TAM in tumour-bearing mice resulted in reduced angiogenesis and decreased
tumour growth and metastatization [130,131]. Moreover, the combination of clodronate with sorafenib, an
available inhibitor of tyrosine protein kinases [e.g. VEGFR and
platelet-derived growth factor receptor (PDGFR)], increased significantly the
efficacy of sorafenib alone in a xenograft model of hepatocellular carcinoma.
In clinical practice, bisphosphonates are employed to treat osteoporosis;
current applications in cancer therapy include their use to treat skeletal
metastases in multiple myeloma, prostate and breast cancer. Treatment with
zoledronic acid was associated with a significant reduction of skeletal-related
events and, possibly, direct apoptotic effects in tumour cells [132–134].
Our
group reported that
the anti-tumour agent of marine origin, trabectedin (Yondelis), was found
unexpectedly to be highly cytotoxic to mononuclear phagocytes, including TAM.
This cytotoxic effect is remarkably selective, as neutrophils and lymphocytes
are not affected [135–137]. Trabectedin has now been registered in 2007 in Europe for the
treatment of soft tissue sarcoma and in 2009 for ovarian cancer [136,138–140].
Another approach is to
inhibit the recruitment of circulating monocytes in tumour tissues. The M-CSF
receptor
(M-CSFR) is expressed exclusively by monocytes–macrophages. In patients with
advanced tumours, clinical studies are under way to check the feasibility and
possibly clinical efficacy of inhibitors to the CSF-1R. Among the many
chemokines expressed in the tumour microenvironment, CCL2 (or monocyte
chemotactic protein-1) occupies a prominent role and has been selected for
therapeutic purposes. Preclinical studies have shown that anti-CCL2 antibodies
or antagonists to its receptor CCR2, given in combination with chemotherapy,
were able to induce tumour regression and yielded to improved survival in mouse
models of prostate cancer or colitis-associated carcinogenesis [141–143].
A third and more recent approach is to ‘re-educate’ TAM to exert
anti-tumour responses protective for the host, ideally by using factors able to
revert TAM into M1-macrophages, with potential anti-tumour activity. It is
becoming accepted that macrophages are flexible and able to switch from one
polarization state to the other [144]. This was achieved in experimental mouse tumours, by injecting
the TLR-9 agonist cytosine–guanine dinucleotide-oligodeoxynucleotide (CpG-ODN),
coupled with anti-IL-10 receptor [145] or the chemokine CCL16 [146]. CpG-ODN also synergized with an agonist anti-CD40 mononuclear
antibody (mAb) to revert TAM displaying anti-tumour activity [147].
A
remarkable anti-tumour
effect of redirected macrophages has been reported recently in human pancreatic
cancer with the use of agonist anti-CD40 mAb [148]. Still in the same direction, a recent report showed that the
plasma protein histidine-rich glycoprotein (HRG), known for its inhibitory
effects on angiogenesis [149,150], is able to skew TAM polarization into M1-like phenotype by
down-regulation of the placental growth factor (PlGF), a member of the VEGF
family. In mice, HRG promoted anti-tumour immune responses and normalization of
the vessel network [151].
The
use of IL-12 in
cancer patients is now under clinical investigation. This cytokine is pivotal
for the simulation of Th1 circuits of adaptive immunity, leading to the
production of IFN-γ. In experimental mouse tumour models, IL-12 injection
reduced the tumour-supportive activities of TAM, suggestive of an M1
polarization [152]. Along the same line, therapies inhibiting IL-6, a main product
of TAM, with specific monoclonal antibodies, may result in reduction of their
M2-skewed phenotype.
Conclusion
The last decade
witnessed a growing understanding of the promoting role of chronic inflammation
in cancer initiation and progression [42,50,51,56,153]. TAM are present
in large numbers in tumour
tissues and are key promoters of cancer-related inflammation [10,11,13–15]. They
produce a host of growth factors and inflammatory cytokines that contribute to
tumour cell survival, development of full-blown angiogenesis and resistance to
therapies. In addition, immunosuppressive mediators released by TAM and related
myeloid cells extinguish host-mediated anti-tumour responses and ease tumour
progression. Therefore, TAM appear to be attractive candidates of future
therapeutic strategies. Depletion of the disloyal TAM in tumours, or their
‘re-education’ to potential anti-tumour effectors, may contribute to increase
the efficacy of current anti-tumour therapies.
[Beyond the scope of this article, and thus
missed is the starvation by fasting and ketogenic diet which target the grossly
defective metabolism of all cancers because of mutations in their
mitochondria. The nearly exclusive
source for ATP, the energy molecule, is through the inefficient fermentation of
glucose. This approach has been able to
cure terminal cancers, over 1000 case histories are in the medical literature,
but the power of pharma has succeeded in blocking clinical trials. I mention
this topic so that (1) this approach,
even if eventually effective, would still not be preferred to a dietary cure.
(2) An approach which weakens the immune system will very likely have
significant side effects arising from limiting the ability to heal and fight
pathogens. The dietary approach is used
with chemotherapy, and thus avoids its side effects, which of course have been
grossly under reported. The only side
effect to fasting is weight loss, and this can be avoided by consuming a high
fat low protein combo, since cancer cells have lost their ability to metabolize
fats, and the proteins would be used for replacement of fast reproducing
cells. Cancer cells wouldn’t have the
ATP for mitosis—jk.]
Acknowledgements
The
Authors are
supported by grants from the Italian Association for Cancer Research (AIRC) and
from Regione Lombardia ‘NEPENTE’ under Institutional Agreement n. 14501A.
Disclosure
The authors have no financial conflict
of interest.
Related content
Articles
related to the one you are viewing
The
articles below have been selected for you based on the article you are
currently viewing.
Authors
Xiaoqiang Tang, Chunfen Mo, Yongsheng Wang,
Dandan Wei, Hengyi
Xiao
Published Date
16 January 2013
Authors
Simon Tazzyman, Claire E. Lewis, Craig Murdoch
Published Date
11 May 2009
Authors
Paola Allavena, Antonio Sica, Cecilia Garlanda,
Alberto
Mantovani
Published Date
19 March 2008
Authors
Michael Allen, J Louise Jones
Published Date
29 October 2010
Authors
Christopher D Gregory, John D Pound
Published Date
25 October 2010
153 of them by clicking on link