Atherosclerosis is an inflammatory disease. Understanding how it develops — from endothelial dysfunction to plaque rupture and heart attack — reveals why the cholesterol hypothesis is incomplete and why inflammation is the key target.
The Stages of Atherogenesis
Stage 1: Endothelial dysfunction
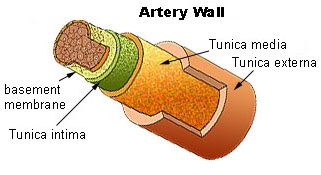
The process begins with damage to the endothelium — the single layer of cells lining the artery. Endothelial dysfunction is caused by:
- Oxidized LDL
- High blood glucose and insulin
- Homocysteine
- Inflammatory cytokines
- Hypertension
Endothelial dysfunction impairs nitric oxide production, causing vasoconstriction and increased permeability to LDL.
Stage 2: LDL entry and oxidation
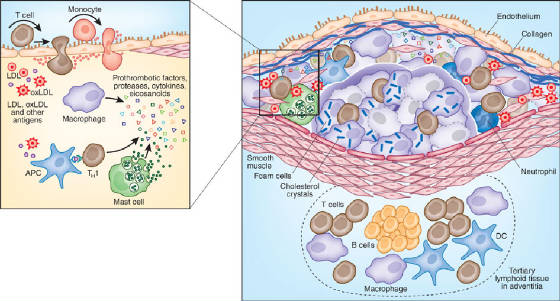
LDL enters the arterial intima through the damaged endothelium. In the intima, LDL is oxidized by reactive oxygen species. Oxidized LDL triggers further inflammation.
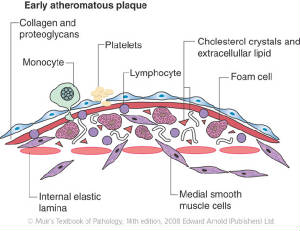
Stage 3: Macrophage recruitment
The endothelium expresses adhesion molecules that recruit monocytes from the blood. Monocytes enter the intima and differentiate into macrophages.
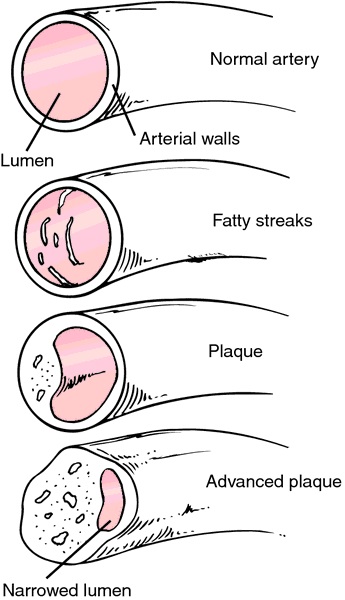
Stage 4: Foam cell formation
Macrophages take up oxidized LDL through scavenger receptors, becoming foam cells. Foam cells accumulate to form the fatty streak — the earliest visible lesion of atherosclerosis.
Atherosclerosis is not a plumbing problem — it is an inflammatory disease. The primary targets for prevention are inflammation and oxidized LDL, not total cholesterol.
Stage 5: Plaque progression
Smooth muscle cells migrate from the media into the intima, proliferating and producing extracellular matrix. The plaque grows, narrowing the arterial lumen.
Stage 6: Plaque rupture
Most heart attacks are not caused by gradual narrowing of the artery. They are caused by rupture of a vulnerable plaque — a plaque with a thin fibrous cap and a large lipid core. When the cap ruptures, the lipid core is exposed to the blood, triggering thrombosis (clot formation) and acute coronary occlusion.
The Inflammatory Hypothesis
The inflammatory hypothesis of atherosclerosis — that inflammation is the primary driver, not cholesterol — is now well-established. Key evidence:
- C-reactive protein (CRP), a marker of inflammation, is a better predictor of cardiovascular events than LDL
- The JUPITER trial showed that rosuvastatin reduced cardiovascular events in people with normal LDL but elevated CRP — suggesting the benefit was anti-inflammatory, not cholesterol-lowering
- Aspirin, which is anti-inflammatory, reduces cardiovascular events more than would be expected from its antiplatelet effects alone
Implications
The primary targets for cardiovascular prevention should be:
- Reducing inflammation (through diet, exercise, and aspirin)
- Reducing LDL oxidation (by eliminating industrial seed oils)
- Reducing endothelial dysfunction (by eliminating fructose and insulin resistance)
- Reducing thrombosis risk (through aspirin and omega-3 fatty acids)